


- Thiết bị máy lạnh
- Phụ kiện nhôm thi công phòng sạch
- Bo kết thúc
- Bo góc ngoài
- Bulong đầu dù 150
- Bo tam giác
- Bo mặt trăng + V nhôm
- Đố cửa đi Panel 50mm
- Đố cửa đi giảm Panel 75mm
- Đố cửa đi giảm Panel 100
- Cây H đi dây điện
- H nối Panel 75mm
- Khung bao kính 1 lớp Panel 50mm
- Khung bao kính 1 lớp Panel 75mm
- Dây cáp treo trần phòng sạch
- T treo trần 60/90/60
- T omega 25/120/25
- Tăng đơ không móc chuyên Bulong dù
- U chân cửa Panel 50mm
- U nhôm 28/50/28
- U nhôm 38/50/28
- U nhôm 38/75/38
- U nhôm 35/100/38
- U nhôm bo 1 bên
- U nhôm bo 2 bên
- V nhôm 40/40
- L nhôm 40/80
- Zoong cao su
- Zoong chân cửa
- Vật liệu panel
- Thế giới cửa
- Cửa panel đơn
- Cửa panel đơn
- Cửa Panel đôi lệch
- Cửa Panel trượt 1 cánh
- Cửa Panel trượt 2 cách
- Cửa panel trượt tự động
- Cửa Panel trượt bán tự động
- Cửa cuốn Nhôm
- Cửa cuốn PVC siêu tốc
- Cửa trượt trần
- Cửa kính trượt tự động
- Cửa Kho lạnh, kho mát
- Cửa kho lạnh tự động
- Cửa cổng xếp tự động
- Cửa nhôm Xingfa
- Cửa nhựa UPVC
- Cửa chống cháy
- Cửa nhựa lõi thép
- Cửa lưới chống muỗi tự động
- Bộ điều khiển HVAC
- Đèn phòng sạch
- Bộ lọc không khí
- Thiết bị Pass Box
- Thiết bị Airshower
- Rèm nhựa PVC
- Sản phẩm điều hoà trane
- Sản phẩm điều hoà Daikin
- Hệ thống Chiller
- Chiller giải nhiệt nước
- Chiller giải nhiệt gió
- Tháp giải nhiệt Cooling Tower
- Thiết bị bơm nước
- Các loại van cơ
- Van cân bằng
- Van điều khiển điện
- Lọc trong hệ thống chiller
- Khớp nối mềm chống rung
- cao su chống rung giảm chấn
- thiết bị đo áp suất
- Thiết bị đo nhiệt độ
- Thiết bị đo Lưu lượng
- công tắc áp suất
- Công tắc dòng chảy
- Thiết bị phụ kiện đường ống
- Thiết kế tủ điện điều khiển AHU khách sạn, biệt thự
- Máy bơm nước công nghiệp
- Tủ điều khiển
- Tủ điều khiển thiết bị AHU dược phẩm
- Tủ điều khiển AHU Phòng mổ bệnh viện
- Tủ điều khiển AHU Phòng điện tử
- Tủ điều khiển AHU khử ẩm sâu
- Tủ điều khiển Chiler
- Tủ điều khiển Cooling Tower
- Tủ điều khiển động cơ
- Tủ điều khiển Kho lạnh Vacxin
- Tủ điều khiển kho lạnh dược phẩm
- Tủ điều khiển kho lạnh thực phẩm
- Tủ điều khiển kho lạnh lưu trữ kem
- Tủ điều khiển bơm
- Tủ điều khiển thiết bị
- Tủ điểu khiển kho lạnh bảo quản nông sản
- Tủ điều khiển BMS
- Tủ điều khiển bảo quản các loại thủy sản
- Tủ điều khiển kho lạnh cấp đông
- Tủ điều khiển kho lạnh vận tải
- Tủ điều khiển hệ thống điều hòa không khí
- Tủ điều khiển FFU
- Tủ điều khiển FCU
- Tủ điều khiển BFU
- Tủ điều khiển quạt hút gió thải
- Tủ điều khiển quạt tăng áp
- Tủ điều khiển Van
- Tủ điều khiển chiếu sáng
- Tủ điều khiển thiết bị AHU
- Sản phẩm quạt thông gió
- Sản phẩm ống gió
- Sàn phòng sạch
- Vật liệu cách nhiệt
- Vật liệu cách âm
- Sơn cách nhiệt
- Phụ kiện tôn gấp
- Thiết bị phòng Lab
- Bảng thí nghiệm
- Clean bench
- Tủ hút khí độc
- Tủ đựng hoá chất
- Tủ cấy vô trùng loại thổi đứng
- Tủ cấy vô trùng loại thổi ngang
- Tủ đựng quần áo sạch
- Tủ an toàn sinh học
- Bàn thí nghiệm áp tường
- Bàn thí nghiệm trung tâm
- Tủ cấy
- Lò sấy sạch
- Kho bảo quản sạch
- Tủ an toàn loại để bàn
- Phòng sạch di động
- Tủ cấy di động dùng ắc quy
- Tủ cấy vô trùng
- Hệ thống sạch trong y tế
- Nội thất y tế inox
- Bồn rửa tay y tế
- Tấm panel phòng mổ
- Hệ thống cấp khí sạch treo trần cho phòng mổ (dòng hai tốc độ)
- Hệ thống cấp khí sạch treo trần cho phòng mổ (dòng một tốc độ)
- Phụ kiện bằng nhựa PVC
- Sàn Vinyl cho bệnh viện
- Cửa kín khí dùng cho phòng mổ
- Hệ thống khí sạch áp lực dương cho phòng mổ
- Mặt bằng phòng mổ
- Hộp van khí y tế
- Hệ thống trạm hút chân không y tế
- Hệ thống máy nén khí y tế
- Thiết bị đo lưu lượng Oxy và điều chỉnh chân không
- Hệ thống khí y tế nhiều đường ống
- Hệ thống cấp oxy cho y tế cấp độ PSA
- Hệ thống khung treo trần y tế
- Khóa Interlock
- SCHNEIDER
- TỦ ĐIỆN ĐỘNG LỰC
- THIẾT XỬ LÝ KHÔNG KHÍ AHU
- Tủ điện phân phối
- Thiết bị tủ điện
Video
Tin mới nhất

Tủ điều khiển AHU - Phòng sạch

Tủ điều khiển Chiller
Chứng nhận tiêu chuẩn GMP là gì ? Phòng sạch GMP

Tiêu chuẩn SSOP là gì? Mối quan hệ giữa GMP, SSOP và HACCP

Bộ điều khiển HVAC (Controller HVAC)

Thiết kế hệ thống thống gió đúng tiêu chuẩn

So sánh 2 phương pháp kiểm tra/thẩm định độ rò rỉ màng lọc HEPA

Quy trình vận hành điều hòa trung tâm VRV

Hướng dẫn chi tiết sử dụng mô hình IO Kit
Quy trình chuẩn bị kiểm tra GMP của bộ y tế Việt Nam
Quy trình chuẩn bị kiểm tra GMP của bộ y tế Việt Nam
1. MỤC ĐÍCH
Mô tả các bước tiến hành chuẩn bị kiểm tra, tiến hành kiểm tra cơ sở đăng ký triển khai GMP để các lần chuẩn bị cho kiểm tra, tiến hành kiểm tra đều được tiến hành theo một trật tự nhất định nhằm:
- Đảm bảo tất cả các đợt chuẩn bị kiểm tra, tiến hành kiểm tra các cơ sở khác nhau đều cùng hiệu quả và cùng một phương pháp
- Công tác kiểm tra luôn tiến hành theo yêu cầu GMP và các quy định hiện hành của Bộ Y tế.
- Mọi thành viên trong Đoàn kiểm tra dễ dàng thực hiện nhiệm vụ.
- Có thể thay đổi khi thiết lập một quy trình mới.
2. PHẠM VI ÁP DỤNG
Áp dụng cho hoạt động kiểm tra cấp giấy chứng nhận GMP của phòng Quản lý chất lượng thuốc của Cục Quản lý Dược.

3. TÀI LIỆU THAM CHIẾU
- Tiêu chuẩn Việt Nam TCVN 9001: 2008 quy định về Hệ thống quản lý chất lượng và các yêu cầu
- Luật Dược ban hành ngày 27/06/2005;
- Quyết định 1570/2000/QĐ-BYT ngày 22/5/2000 ban hành nguyên tắc, tiêu chuẩn "Thực hành tốt phòng kiểm nghiệm thuốc" của Bộ Y tế;
- Quyết định số 2701/2001/QĐ-BYT ngày 29/6/2001 ban hành nguyên tắc, tiêu chuẩn "Thực hành tốt bảo quản thuốc" của Bộ trưởng Bộ Y tế;
- Quyết định 3886/2004/QĐ-BYT ngày 3/11/2004 ban hành nguyên tắc, tiêu chuẩn "Thực hành tốt sản xuất thuốc" theo khuyến cáo của Tổ chức y tế thế giới;
- Quyết định 27/2007/QĐ-BYT ngày 19/4/2007 về việc ban hành lộ trình triển khai áp dụng các nguyên tắc, tiêu chuẩn “Thực hành tốt sản xuất thuốc" và nguyên tắc “Thực hành tốt bảo quản thuốc";
- Quyết định 47/2007/QĐ-BYT ngày 24/12/2007 về việc triển khai áp dụng các nguyên tắc, tiêu chuẩn “Thực hành tốt sản xuất thuốc", “Thực hành tốt phòng kiểm nghiệm thuốc", nguyên tắc “Thực hành tốt bảo quản thuốc" và nguyên tắc “Thực hành tốt phân phối thuốc" đói với các cơ sở sản xuất, kiểm nghiệm, kinh doanh, phân phối, xuất khẩu, nhập khẩu, tốn trữ, bảo quản vắc xin và sinh phẩm y tế.
- Thông tư số 45/2011/TT-BYT ngày 21/12/2011 của Bộ Y tế sửa đổi, bổ sung một số điều của Quyết định số 1570/2000/QĐ-BYT ngày 22/5/2000 của Bộ trưởng Bộ Y tế về việc triển khai áp dụng nguyên tắc “Thực hành tốt phòng kiểm nghiệm thuốc”; Quyết định số 2701/2001/QĐ-BYT ngày 29/6/2001 của Bộ trưởng Bộ Y tế về việc triển khai áp dụng nguyên tắc “Thực hành tốt bảo quản thuốc”; Thông tư số 06/2004/TT-BYT ngày 28/5/2004 hướng dẫn sản xuất gia công thuốc; Quyết định 3886/2004/QĐ-BYT ngày 03/11/2004 của Bộ Y tế về việc triển khai áp dụng nguyên tắc, tiêu chuẩn “Thực hành tốt sản xuất thuốc” theo khuyến cáo của Tổ chức Y tế thế giới; Thông tư số 13/2009/TT-BYT ngày 01/9/2009 của Bộ Y tế hướng dẫn hoạt động thông tin quảng cáo thuốc; Thông tư số 22/2009/TT-BYT ngày 24/11/2009 của Bộ Y tế quy định về đăng ký thuốc; Thông tư số 47/2010/TT-BYT ngày 29/12/2010 hướng dẫn hoạt động xuất khẩu, nhập khẩu thuốc và bao bì tiếp xúc trực tiếp với thuốc
- Công văn số 8071/QLD-CL ngày 15/10/2004 về việc triển khai đồng thời GMP, GLP, GSP của Cục trưởng Cục Quản lý Dược.
- Quyết định 192/QLD-CL ngày 10/04/2014 của Cục trưởng Cục Quản lý Dược về việc ban hành danh sách chuyên gia thẩm định hồ sơđăng ký kiểm tra GMP, GLP và GSP.
- Quyết định 193/QLD-CL ngày 10/04/2015 của Cục trưởng Cục Quản lý Dược về việc ban hành danh sách thanh tra viên kiểm tra GMP, GLP và GSP.
4. TRÁCH NHIỆM THỰC HIỆN
- Lãnh đạo Cục có trách nhiệm kiểm tra và bảo đảm những quy định trong quy trình này được thực hiện và tuân thủ.
- Lãnh đạo Phòng có liên quan đến quy trình có trách nhiệm phối hợp, kiểm tra và bảo đảm những quy định trong quy trình này được thực hiện và tuân thủ.
- Chuyên viên liên quan đến quy trình có trách nhiệm thực hiện và tuân thủ những quy định trong quy trình này.
5. ĐỊNH NGHĨA VÀ CHỮ VIẾT TẮT
- GMP: Thực hành tốt sản xuất thuốc.
- GLP: Thực hành tốt phòng kiểm nghiệm thuốc
- GSP: Thực hành tốt bảo quản thuốc
- SOP: Quy trình chuẩn
- Phòng QLCL thuốc: Phòng Quản lý chất lượng thuốc.
6. NỘI DUNG QUY TRÌNH
6.1. Sơ đồ quá trình quy trình chuẩn bị kiểm tra GMP của bộ y tế Việt Nam
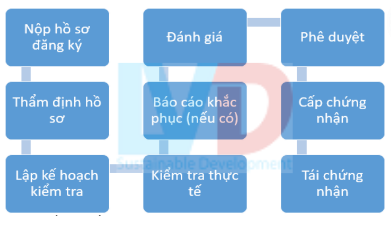
6.2. Kiểm tra GMP thường kỳ
6.2.1.Tiếp nhận hồ sơ đăng ký
* Thời gian tối đa thực hiện: 2 ngày.
- Hồ sơ đăng kýkiểm tra GMP kèm theo Biên nhận nộp phí được tiếp nhận tại Bộ phận văn thư/bộ phận một cửa - Văn phòng Cục. Hồ sơ kiểm tra được kiểm tra theo Checklist hồ sơ đăng ký kiểm tra GMP lần đầu đối với cơ sở đăng ký lần đầu (BM.CL.01/01) và Checklist hồ sơ đăng ký tái kiểm tra GMP đối với cơ sở tái đăng ký (BM.CL.01/02). Văn thư Cục vào sổ theo dõi văn bản đến với số văn bản đến.
- Sau khi đơn vị hoàn tất thủ tục tài chính, trong vòng 01 ngày Văn phòng Cục chuyển Hồ sơ đăng ký kiểm tra cho Phòng QLCL thuốc.
- Trong vòng 1 ngày, văn thư Phòng QLCL thuốc sau khi tiếp nhận hồ sơ, phải vào sổ nhận công văn đến của Phòng, chuyển Lãnh đạo Phòng phân công và chuyển đến chuyên viên thụ lý hồ sơ.
- Trường hợp tiếp nhận hồ sơ đề nghị cấp Giấy đủ điều kiện Kinh doanh dược liên thông từ Phòng Quản lý Kinh doanh dược, sau khi được Lãnh đạo phòng phân công, chuyên viên thụ lý hồ sơ xử lý theo trình tự như các hồ sơ đăng ký GMP được tiếp nhận từ Văn phòng Cục.
- Chuyên viên thụ lý hồ sơcập nhật vào Danh sách nhà máy đăng ký kiểm tra GPs (theobiểu mẫu bảng Excel BM.CL.01/03).
6.2.2.Thẩm định hồ sơ
* Thời gian tối đa thực hiện: 3 ngày.
a) Chuẩn bị
Chuyên viên thụ lý hồ sơ lập biên bản thẩm định theo các biểu mẫu dưới đây, chuyển hồ sơ, biên bản thẩm định đến các chuyên gia thẩm định và Lãnh đạo Phong xem xét, kết luận.
* Mẫu biên bản thẩm định:
+ Biên bản thẩm định hồ sơ đăng ký kiểm tra GMP lần đầu: BM.CL.01/04
+ Biên bản thẩm định hồ sơ tái đăng ký kiểm tra GMP: BM.CL.01/05
* Chuyên gia thẩm định:
Chuyên viên phòng QLCL thuốc và các phòng liên quan theo Quyết định thành lập của Cục trưởng Cục Quản lý Dược.
b) Thẩm định hồ sơ
+ Đối với hồ sơ đăng ký kiểm tra lần đầu: chuyên gia phải đánh giá sự phù hợp về quy mô và tính hợp lý của việc triển khai tại cơ sở với các dây chuyền cơ sở đăng ký kiểm tra (cấp sạch nhà xưởng; công suất thiết bị, hệ thống; chiều di chuyển của nhân viên, nguyên liệu, bán thành phẩm, thành phẩm; phân công và trình độ của nhân sự chủ chốt;...) và các điểm cần lưu ý trong quá trình kiểm tra (các nội dung chưa phù hợp hoặc nghi ngờ...)
+ Đối với hồ sơ đăng ký tái kiểm tra: chuyên gia phải xem xét đánh giá kết quả kiểm tra lần trước về mức độ đáp ứng yêu cầu GMP, các thay đổi so với lần kiểm tra trước (các ảnh hưởng của việc thay đổi đến điều kiện môi trường, thiết bị, quy trình,...), báo cáo khắc phục các tồn tại của lần kiểm tra trước (việc thực hiện các tồn tại theo kế hoạch xây dựng được báo cáo gần nhất), các chú ý liên quan đến vi phạm quy định hiện hành về Dược của cơ sở (nếu có): vi phạm về chất lượng thuốc, sản xuất thuốc,....
c) Kết quả thẩm định và xem xét kết quả thẩm định
- Trường hợp hồ sơ đáp ứng đầy đủ quy định, chuyên viên thụ lý phải:
+ Ghi kết quả thẩm định vào Danh sách nhà máy đăng ký kiểm tra GPs (biểu mẫu BM.CL.01/03) theo dõi tình trạng hồ sơ.
+ Thông báo chính thức cho cơ sở (qua văn thư/email/fax/tin nhắn,...), thống nhất thời gian dự kiến kiểm tra phù hợp với hoạt động của cơ sở.
- Trường hợp hồ sơ chưa đáp ứng đầy đủ theo quy định:
+ Ghi kết quả thẩm định vào Danh sách nhà máy đăng ký kiểm tra GPs (biểu mẫu BM.CL.01/03) theo dõi tình trạng hồ sơ.
+ Thông báo chính thức cho cơ sở (qua văn thư/email/fax/tin nhắn,...) nêu rõ các nội dung cần bổ sung. Đối với hồ sơ tái đăng ký, thời gian bổ sung không quá 30 ngày hoặc đến thời điểm hết hiệu lực của giấy chứng nhận GMP.
Khi nhận được hồ sơ bổ sung, chuyên viên thụ lý hồ sơ chịu trách nhiệm xem xét hồ sơ bổ sung, báo cáo Lãnh đạo Phòng để kết luận.
Trường hợp đối với hồ sơ tái đăng ký không được bổ sung đúng thời hạn, chuyên viên thụ lý hồ sơ ghi rõ trong biên bản thẩm định, chuyển hồ sơ tới Trưởng phòng xem xét để đề xuất và báo cáo Lãnh đạo Cục biện pháp xử lý.
6.2.3.Xây dựng kế hoạch và thành lập Đoàn kiểm tra
* Thời gian tối đa thực hiện:3 ngày.
a) Tuần 1 hàng tháng, Lãnh đạo Phòng thảo luận và phân công chuyên viên chuẩn bị kế hoạch kiểm tra GPs, dự thảo các Quyết định thành lập đoàn kiểm tra, trình Lãnh đạo Cục xem xét:
- Kế hoạch kiểm tra bao gồm:
+ Danh sách các cơ sở nộp hồ sơ đăng ký đạt yêu cầu.
+ Lịch trình kiểm tra dự kiến
+ Thành phần đoàn kiểm tra
- Quyết định thành lập Đoàn kiểm tra
- Thành phần đoàn kiểm tra:
+ Trưởng đoàn: Lãnh đạo Cục, Lãnh đạo phòng Quản lý Chất lượng thuốc hoặc Lãnh đạo phòng khác theo sự phân công của Cục trưởng;
+ Thư ký đoàn: Chuyên viên phòng Quản lý chất lượng thuốc;
+ Các thành viên: Sở Y tế địa phương và có thể thêm đại diện phòng chức năng khác của Cục Quản lý Dược;
- Thời gian kiểm tra: Dự kiến 01 - 02 ngày.
b) Lãnh đạo Cục xem xét kế hoạch kiểm tra:
- Nếu đồng ý, Lãnh đạo Cục phê duyệt kế hoạch kiểm tra tháng và ký ban hành các Quyết định thành lập Đoàn kiểm tra và chuyển trả Văn thư Cục.
Văn thư Cục tiến hành thủ tục ban hành các Quyết định thành lập Đoàn kiểm tra; lưu 01 bản và gửi trả Phòng QLCL kèm kế hoạch kiểm tra.
- Nếu không đồng ý, Lãnh đạo Cục cho ý kiến điều chỉnh kế hoạch kiểm tra, thành phần các đoàn kiểm tra và trả lại Phòng QLCL để thực hiện lại từ bước a.
6.2.4.Chuẩn bị thực hiện kiểm tra cơ sở
Thời gian tối đa thực hiện: 2 ngày.
a) Chuyên viên thụ lý hồ sơ bàn giao hồ sơ đăng ký, biên bản thẩm định và các tài liệu liên quan cho thư ký đoàn kiểm tra.
b) Thư ký đoàn:
+ Chuyển Quyết định kiểm tra đến các thành viên của đoàn kiểm tra.
+ Chuyển Quyết định kiểm tra và thông báo thời gian kiểm tra tại cơ sở theo kế hoạch đã được phê duyệt.
c) Xây dựng chương trình kiểm tra
Ít nhất 01 ngày, trước thời điểm tiến hành kiểm tra tại cơ sở, Thư ký đoàn dự thảo chương trình cụ thể về kiểm tra GMP tại cơ sở (theo biểu mẫu BM.CL.01/06).
Chương trình kiểm tra cần phải đảm bảo phù hợp với đặc thù của từng đơn vị, đáp ứng mục đích và thời gian của đợt kiểm tra. Kế hoạch kiểm tra phải căn cứ trên dây chuyền đăng ký kiểm tra đối với cơ sở mới, trên kết quả kiểm tra lần trước và các hành động khắc phục sau đợt kiểm tra đối với cơ sở tái kiểm tra, các thông tin về các vi phạm của cơ sở (nếu có). Kế hoạch kiểm tra phải bao gồm các khu vực và thời gian dự kiến kiểm kiểm tra của từng khu vực, danh mục các tài cần phải kiểm tra (có thể là tài liệu riêng, đính kèm kế hoạch kiểm tra) và đầy đủ các thông tin khác theo biểu mẫu.
Chương trình kiểm tra có thể được thay đổi trong quá trình thanh tra nếu phát hiện các điểm cần kiểm tra kỹ hơn hoặc mở rộng đối tượng, phạm vi kiểm tra. Khi thực hiện, cần xem xét tiến trình kiểm tra, các phát hiện trong qusa trình kiểm tra, đối chiếu với chương trình kiểm tra và điều chỉnh nếu cần.
Trưởng đoàn chịu trách nhiệm tổ chức họp đoàn kiểm tra trước khi tiến hành kiểm tra để rà soát phân công trách nhiệm của các thành viên, chuẩn bị nội dung cần chú ý trong kiểm tra và thống nhất chương trình kiểm tra tại cơ sở sản xuất và thông báo chương trình kiểm tra cho cơ sở sản xuất.
6.2.5.Kiểm tra tại cơ sở
* Thời gian tối đa thực hiện: 5 ngày.
6.2.5.1. Trình tự kiểm tra
a) Đoàn kiểm tra, theo hướng dẫn của đại diện cơ sở, đi một vòng bên ngoài, đánh giá sơ bộ về cơ sở sản xuất.
b) Họp khai mạc với cơ sở
-Đại diện đoàn kiểm tra: Công bố Quyết định, giới thiệu thành phần đoàn kiểm tra, mục đích, phạm vi, phương pháp, trình tự và chương trình kiểm tra.
-Đại diện Cơ sở giới thiệu thành phần tham dự họp, báo cáo tóm tắt về hoạt động và việc triển khai áp dụng GMP(thời gian báo cáo không quá 60 phút) với các nội dung sau:
* Đối với cơ sở kiểm tra mới:
+ Giới thiệu khái quát về cơ sở: Giới thiệu hoạt động chung của cơ sở, Sơ đồ tổ chức
+ Tóm tắt về quá trình đào tạo và kết quả đào tạo GMP
+ Sơ đồ các khu vực sản xuất: Sơ đồ mặt bằng địa lý, Sơ đồ bố trí dây truyền sản xuất: bố trì phòng sản xuất, cấp sạch, chênh lệch áp xuất..., Đường di chuyển của công nhân, của nguyên vật liệu, bán thành phẩm, thành phẩm…
+ Các hệ thống phụ trợ: Hệ thống xử lý không khí; Hệ thống cung cấp nước sản xuất; hệ thống xử lý chất thải rắn, chất thải lỏng, khí thải;Hệ thống phòng cháy, chữa cháy và an toàn lao động,...
+ Tình hình hoạt động sản xuất: Dạng bào chế cơ sở sản xuất đăng ký kiểm tra, Kiểm tra trong quá trình sản xuất, Đánh giá nhà cung cấp, Các hoạt động thẩm định,...
+ Hoạt động của phòng kiểm tra chất lượng: theo quy định về GLP.
+ Hoạt động của bảo quản thuốc: theoquy định về GSP.
+ Thanh tra nội bộ.
Trong quá trình trình bày hoặc kết thúc bài giới thiệu của cơ sở, các thành viên đoàn kiểm tra nêu các câu hỏi về những điều cần làm rõ trong hồ sơ đăng ký và trong nội dung báo cáo của cơ sở nhằm tìm hiểu thêm các thông tin cần thiết về cơ sở trước khi đoàn kiểm tra thực tế.
*Đối với cơ sở tái kiểm tra:
+ Báo cáo hoạt động của cơ sở và các thay đổi của cơ sở trong 3 năm.
+ Báo cáo khắc phục các tồn tại trong lần kiểm tra trước.
+ Báo cáo về việc đào tạo cán bộ trong 3 năm.
c) Kiểm tra thực tế các hoạt động của cơ sở
Đoàn kiểm tra thực hiện việc kiểm tra thực tế:
- Kiểm tra trực tiếp tại các khu vực sản xuất, kiểm nghiệm, kho bảo quản nguyên liệu, thành phẩm, bao bì, các hệ thống phụ trợ,...
- Kiểm tra rà soát hồ sơ lưu trữ về các hoạt động của cơ sở sản xuất.
Ghi chú: Thông thường, đoàn kiểm tra tiến hành kiểm tra trực tiếp tại các khu vực trước khi kiểm tra về hồ sơ được lưu trữ. Tuy nhiên, trong quá trình kiểm tra, rà soát hồ sơ lưu, thanh tra viên có thể tiến hành kiểm tra trực tiếp lại tại khu vực thực hiện hoạt động đó.
Các thanh tra viên ghi lại các dữ liệu từ việc đặt câu hỏi hoặc phỏng vấn trực tiếp tới người quản lý hoặc nhân viên vận hành, xem xét các hồ sơ, quan sát quá trình vận hành,...
- Thành viên đoàn kiểm tra phải ghi lại vào Bản ghi chép kiểm tra (biểu mẫuBM.CL.01/07) hoặc sổ tay thanh tra viên các quan sát, các tài liệu đã xem và thông báo những điểm không phù hợp cho nhân viên của cơ sở sản xuất trước khi rời khỏi khu vực đã kiểm tra. Việc ghi chép cần phải sử dụng bút bi, bút mực, không sử dụng bút chì. Thanh tra viên phải ghi lại và đưa vào báo cáo kiểm tra/biên bản kiểm tra tất cả các điểm không phù hợp phát hiện được, kể cả các điểm mà nhà sản xuất đã khắc phục ngay sau đó.
- Trong quá trình kiểm tra, thanh tra viên có thể lấy mẫu nguyên liệu, bán thành phẩm, thành phẩm để gửi đi phân tích, kiểm nghiệm nếu nhận thấy có dấu hiệu nghi ngờ về chất lượng.
- Thanh tra viên có quyền được tiếp cận tất cả các khu vực sản xuất, kiểm tra chất lượng, bảo quản, các hệ thống phụ trợ và các hồ sơ tài liệu liên quan đến các hoạt động sản xuất, kiểm tra chất lượng, bảo quản thuốc, bao gồm các SOP, các đề cương, sơ đồ, bản ghi chép, các dữ liệu và hệ thống máy tính. Trong trường hợp cần thiết, thanh tra viên có thể yêu cầu cung cấp bản photocopy tài liệu hoặc chụp ảnh, quay video cơ sở vật chất, thiết bị, nhà xưởng...
- Trong trường hợp phát hiện vi phạm ảnh hưởng nghiêm trọng tới chất lượng của một sản phẩm hoặc nhiều sản phẩm; trưởng đoàn cần phải lập biên bản, đánh giá nguy cơ ảnh hưởng của vi phạm và yêu cầu cơ sở tạm ngừng hoạt động sản xuất liên quan đến vi phạm; yêu cầu ngừng này phải được thông báo lại trong buổi họp kết thúc kiểm tra và phải được ghi vào trong biên bản/báo cáo kiểm tra).Biên bản này phải được báo cáo Lãnh đạo Cục để biết và có văn bản xử lý chính thức.
- Việc họp đoàn kiểm tra, tóm tắt các hoạt động kiểm tra đã thực hiện, các vướng mắc trong quá trình kiểm tra và các các tồn tại phát hiện được của từng ngày có thể được thực hiện vào cuối ngày kiểm tra hoặc đầu ngày hôm sau. Trưởng Đoàn kiểm tra tập hợp ý kiến của các thành viên, lập danh sách các điểm không phù hợpcủa từng ngày kiểm tra và thông báo cho cơ sở sản xuất. Trường hợp cơ sở không thống nhất với các nội dung phát hiện, cơ sở phải chuẩn bị và cung cấp các bằng chứng cho đoàn vào ngày kiểm tra kế tiếp.
c) Họp đoàn kiểm tra cuối đợt kiểm tra
Đoàn kiểm tra sẽ họp riêng để thống nhất ý kiến; thư ký đoàn chịu trách nhiệm tập hợp các ý kiến, các phát hiện trong quá trình kiểm tra của các thành viên,tổng hợp, thống nhất danhsách các tồn tại, phân loại các tồn tại (Theo Phụ lục I) và dự kiến kết luận về mức độ tuân thủ GMP của cơ sở kiểm tra.
Đoàn kiểm tra họp với cơ sở, thông báo tóm tắt quá trình kiểm tra, các tồn tại phát hiện, phân loại tồn tại. Cơ sở có thể giải thích, thảo luận đối với các vấn đề mà cơ sở chưa rõ, hoặc chưa thống nhất với đánh giá của đoàn kiểm tra. Tất cả các ý kiến không thống nhất của cơ sở phải được ghi lại và đưa vào nội dung của Biên bản kiểm tra.
Trưởng đoàn thông báo đánh giá mức độ tuân thủ dự kiến của cơ sở.
A - Cơ sở tuân thủ tốt GMP: Cơ sở không có tồn tại nghiêm trọng hay tồn tại nặng nào.
B - Cơ sở tuân thủ GMP: Cơ sở không có tồn tại nghiêm trọng nào, có từ 1 đến 6 tồn tại nặng.
C - Cơ sở tuân thủ GMP ở mức cơ bản: Cơ sở không có tồn tại nghiêm trọng nào và có từ 7-14 tồn tại nặng.
D - Cơ sở không tuân thủ GMP: Nếu cơ sở cónhiều hơn hoặc bằng 1 tồn tại nghiêm trọng và/hoặc có từ 15 tồn tại nặng trở lên.
* Đoàn kiểm tra lập Danh sách thành phần tham dự đợt kiểm tra (biểu mẫu BM.CL.01/09); Bản đánh giá sơ bộ kết quả đợt kiểm tra (biểu mẫu BM.CL.01/16). Trưởng đoàn kiểm tra và Đại diện cơ sở ký tên.
6.2.6.Hoàn thiện biên bản kiểm tra
Thời gian tối đa thực hiện: 1 tháng (Trường hợp cụ thể theo mục 6.2.7).
- Đoàn kiểm tra (Trưởng đoàn và Thư ký) hoàn thiện Biên bản kiểm tra GMP (theo BM.CL.01/08),
- Biên bản kiểm tra phải được mô tả chi tiết đối với nhà xưởng, hệ thống, nhân sự, tình hình triển khai từng nội dung theo yêu cầu của nguyên tắc,... các nội dung đã được kiểm tra, các hồ sơ tài liệu đã được xem xét, các dạng sản phẩm đang được tiến hành trong quá trình kiểm tra,...Đối với tồn tại, phải mô tả cụ thể tình trạng, địa điểm, thời gian và các yếu tố ảnh hưởng (nếu có) để đưa đến việc đánh giá mức độ của tồn tại; được liệt kê, xếp loại và tham chiếu đến các điều, khoản tại các tài liệu hướng dẫn về “GMP” của WHO, PICs hoặc các văn bản quy phạm liên quan.
- Thư ký Đoàn trình Trưởng phòng QLCL rà soát, nếu Trưởng phòng tham gia Đoàn kiểm tra thì chuyển Phó Trưởng phòng rà soát biên bản trước khi ký với cơ sở. Người rà soát ký tên vào bản dự thảo của biên bản kiểm tra.
- Biên bản kiểm tra GMP sau khi được rà soát, hoàn thiện và gửi cho cơ sở sản xuất.
Đối với cơ sở phải báo cáo khắc phục, biên bản kiểm tra nêu rõ thời gian yêu cầu cơ sở gửi báo cáo khắc phục, kế hoạch khắc phục về Cục Quản lý Dược.
- Thư ký đoàn kiểm tra chịu trách nhiệm lưu giữ biên bản kiểm tra và tiếp nhận, xử lý đối với báo cáo khắc phục tồn tại của cơ sở sản xuất, dự thảo các Phiếu trình, các Quyết định cấp và Giấy chứng nhận GMP.
6.2.7.Xử lý sau khi kiểm tra
Việc xử lý kết quả kiểm tra sẽ tùy theo vào mức độ tuân thủ GMP của cơ sở:
a) Cơ sở tuân thủ tốt GMP (A)
Chuyển sang bước 6.2.8: Trình lãnh đạo Cục cấp Chứng nhận GMP.
b) Cơ sở tuân thủ GMP (B)
* Thời gian thực hiện tối đa: 5 ngày
Cơ sở phải xây dựng Kế hoạch khắc phục và gửi về Cục Quản lý Dược trong vòng 2 tháng kể từ ngày kết thúc đợt kiểm tra.
Trong vòng 5 ngày kể từ khi nhận được Kế hoạch khắc phục của cơ sở, Trưởng đoàn và Thư ký đoàn chịu trách nhiệm đánh giá tính phù hợp của kế hoạch khắc phục:
+ Nếu Kế hoạch khắc phục phù hợp, chuyển sang bước 6.2.8: Trình lãnh đạo Cục cấp Chứng nhận GMP.
+ Trường hợp kế hoạch khắc phục chưa phù hợp, thư ký Đoàn thông báo cho cơ sở (qua văn thư/email/tin nhắn,....) để hoàn thiện Kế hoạch và quay lại đầu bước 6.2.7b.
c) Cơ sở tuân thủ GMP ở mức cơ bản (C)
* Thời gian thực hiện tối đa: 10 ngày
Cơ sở phải gửi báo cáo khắc phục những điểm không phù hợp về Cục Quản lý Dược trong vòng 2 tháng kể từ ngày kết thúc đợt kiểm tra. Báo cáo khắc phục bao gồm Kế hoạch khắc phục và các bằng chứng, tài liệu chứng minh đối với các hành động đã khắc phục.
Trong vòng 5 ngày kể từ khi nhận được Báo cáo khắc phục của cơ sở, Đoàn kiểm tra (Trưởng đoàn và Thư ký) tiến hành đánh giá báo cáo khắc phục của cơ sở theo biểu mẫu đánh giá báo cáo khắc phục (BM.CL.01/12); chuyển Trưởng phòng báo cáo Lãnh đạo Cục cho ý kiến về kết luận mức độ đáp ứng của cơ sở:
+Nếu Báo cáo khắc phục đã đáp ứng yêu cầu (số tồn tại nặng chưa khắc phục được dưới 6 và cơ sở có kế hoạch cụ thể để khắc phục những tồn tại này), chuyển sang bước 5.2.8: Trình lãnh đạo Cục cấp Chứng nhận GMP.
+ Trường hợp báo cáo khắc phục của cơ sở chưa đáp ứng yêu cầu (số tồn tại nặng chưa khắc phục được lớn hơn 6), thư ký Đoàn thông báo cho cơ sở (qua văn thư/email/tin nhắn,....) để tiếp tục hoàn thiện Báo cáo khắc phục, trong đó đánh giá mức độ khắc phục, những điểm chưa khắc phục được và yêu cầu cơ sở phải nộp báo cáo khắc phục bổ sung trong vòng 02 tháng tiếp theo và quay lại đầu bước 6.2.7c.
Cơ sở được báo cáo khắc phục tối đa 2 lần. Nếu ngoài thời gian 2 tháng, cơ sở chưa nộp báo cáo khắc phục hoặc sau 2 lần báo cáo khắc phục vẫn không đáp ứng yêu cầu, chuyển bước d).
d) Cơ sở không tuân thủ nguyên tắc GMP
Cục Quản lý Dược kết luận cơ sở không tuân thủ GMP và có Quyết định yêu cầu cơ sở phải dừng ngay việc sản xuất (Biểu mẫu BM.CL.01/13).Cơ sở phải nộp lại hồ sơ đăng ký kiểm tra GMP sau khi hoàn thành các hành động khắc phục. Cơ sở chỉ được phép tiếp tục sản xuất sau khi được kiểm tra và cấp giấy chứng nhận GMP.
Đối với trường hợp, sự không tuân thủ chỉ ảnh hưởng tới một phần của phạm vi chứng nhận, Đoàn kiểm tra sẽ đề xuất với Lãnh đạo Cục về việc sửa đổi, thu hẹp phạm vi chứng nhận.
đ) Trường hợp đặc biệt
Trong một số trường hợp dưới đây, phòng QLCL sẽ đề xuất lãnh đạo Cục Quản lý Dược/Bộ Y tế cân nhắc nguy cơ và lợi ích, để ra quyết định có cho phép cơ sở được đánh giá là không tuân thủ GMP tiếp tục sản xuất một hoặc một số sản phẩm xác định trong một khoảng thời gian xác định:
+ Cung ứng thuốc, vắc xin phục vụ nhu cầu cấp bách cho phòng, chống dịch bệnh, khắc phục hậu quả, thiên tai, thảm họa;
+ Nguồn cung cấp sản phẩm thay thế không có, không đủ hoặc không kịp đáp ứng;
+ Có các biện pháp hiệu quả nhằm tăng cường kiểm tra, giám sát để đảm bảo hoặc giảm thiểu nguy cơ về chất lượng và kịp thời xử lý khi có diễn biến bất lợi.
Trường hợp cơ sở được đánh giá là cơ sở không tuân thủ GMP và có Quyết định yêu cầu cơ sở phải dừng ngay việc sản xuất, cơ sở sau khi được kết luận không tuân thủ GMP để tiếp tục sản xuất một hoặc một số sản phẩm xác định trong một khoảng thời gian xác định hoặc cơ sở bị rút chứng nhận GMP, sau khi hoàn tất các thủ tục trên, thư ký đoàn cập nhật vào danh sách các đơn vị bị dừng sản xuất, rút giấy chứng nhận GMP.
6.2.8.Trình Lãnh đạo Cục cấp chứng nhận GMP
Thời gian tối đa thực hiện: 5 ngày.
Trong vòng 5 ngày kể từ ngày kiểm tra (đối với cơ sở Tuân thủ tốt - A) hoặc kể từ ngày hoàn thành các bước xử lý sau kiểm tra (đối với cơ sở Tuân thủ - B; hoặc Tuân thủ ở mức cơ bản - C), thư ký đoàn báo cáo trưởng phòng Quản lý chất lượng thuốc, dự thảo các Phiếu trình Lãnh đạo Cục để ban hành Quyết định cấp giấy chứng nhận (Biểu mẫu BM.CL.01/10) và Giấy chứng nhận GMP (BM.CL.01/11). Đoàn kiểm tra trình, giải thích và làm rõ các nội dung liên quan nếu có yêu cầu nào của Lãnh đạo Cục.
Đối với hồ sơ đề nghị cấp Giấy đủ điều kiện Kinh doanh dược liên thông từ Phòng Quản lý Kinh doanh dược, sau khi Cục trưởng ký ban hành Quyết định và Giấy chứng nhận, Thư ký đoàn gửi công văn, kèm theo bản photocopy của Biên bản kiểm tra, Quyết định cấp và Giấy Chứng nhận đến Phòng Quản lý Kinh doanh dược để hoàn tất các thủ tục tiếp theo.
6.3. Kiểm tra giám sát
Tiến hành như nội dung tại mục 6.2.3, 6.2.4, 6.2.5, 6.2.6, 6.2.7 của phần kiểm tra thường kỳ trong đó tập trung vào nội dung báo cáo khắc phục của Cơ sở.
6.4. Kiểm tra đột xuất
- Tiến hành như nội dung tại mục 6.2.3, 6.2.4, 6.2.5, 6.2.6, 6.2.7 của phần kiểm tra thường kỳ trong đó tập trung vào lý do của việc kiểm tra đột xuất.
- Lãnh đạo phòng QLCL thuốcdự kiến thành phầnĐoàn kiểm tra và thời gian kiểm tra, trình Lãnh đạo Cục ký ban hành quyết định kiểm tra. Quyết định được gửi tới cơ sở sản xuất trong vòng 24-48h. Trường hợp cần thiết Quyết định có thể đươc gửi tới cơ sở ngay tại thời điểm đến.
- Phụ thuộc vào lý do dẫn đến kiểm tra đột xuất, việc kiểm tra sẽ được đánh giá theo sản phẩm (có tính cá biệt) hay đánh giá theo quy trình (có tính hệ thống). Bao gồm:
* Căn cứ theo sản phẩm : tập trung vào quá trình phát triển của sản phẩm từ nguyên liệu ban đầu tới giai đoạn đóng gói cấp 2, bao gồm các nội dung sau:
+ Tiêu chuẩn của nguyên liệu ban đầu và bao bìđóng gói;
+ Các SOP;
+ Lược đồ sản xuất/ hồ sơ sản xuất;
+ Nhật ký thiết bị;
+ Quy trình lấy mẫu;
+ IPC các sản phẩm trung gian và sản phẩm chờđóng gói;
+ Quy trình kiểm nghiệm/ hồ sơ;
+ Quy trình xuất xưởng
* Căn cứ theo quy trình:
+ Quy trình thay trang phục đi của nhân viên ra/vào phòng sạch;
+ Lấy mẫu nguyên liệu;
+ Quy trình vệ sinh thiết bị;
+ Vận chuyển sản phẩm thải loại;
+ Quy trình đóng gói các lô có kích cỡ nhỏ;
+ Quy trình cân nguyên liệu ban đầu;
+ Thử nghiệm tính toàn vẹn màng lọc trong quá trình sản xuất sản phẩm vô trùng.
* Căn cứ theo khu vực sản xuất : tập trung vào vùng có nguy cơ cao:
+ Tình trạng vệ sinh và làm sạch của phòng sản xuất và nhà xưởng;
+ Tình trạng của trần, tường, sàn;
+ Nhãn tình trạng của nguyên liệu, thiết bị, các đường ống;
+ Khu vực tạm trữ vàđường đi nguyên liệu;
+ Nhật ký thiết bị (hiệu chuẩn, bảo trì bảo dưỡng, sự cố);
+ Phòng tránh nhiễm chéo;
+ Yêu cầu về cấu trúc hoặc hồ sơ thực địa;
+ Quy định về trách nhiệm ở khu vực sản xuất;
+ Việc mặc trang phục vàthao tác của nhân viên, vị trílàm việc trong khu vực sản xuất;
+ Tài liệu tập huấn nhân viên;
6.5. Xếp loại nguy cơ và tần suất kiểm tra giám sát tuân thủ GMP
Việc xếp loại nguy cơ của cơ sở sản xuất là một nội dung của việc kiểm tra GMP sử dụng công cụ quản lý rủi ro. Xếp loại nguy cơ cơ sở sản xuất căn cứ trên việc đánh giá 2 loại nguy cơ khác nhau: nguy cơ nội tại và nguy cơ liên quan tới việc tuân thủ GMP.
Nguy cơ nội tại của cơ sở sản xuất phản ánh mức độ phức tạp của cơ sở, quy trình sản xuất, dạng sản phẩm cũng như mức độ ảnh hưởng/ nguy cơ của sản phẩm hoặc các hoạt động của cơ sở bao gồm cả khía cạnh cung cứng.
Nguy cơ liên quan tới việc tuân thủ GMP phản ánh tình trạng tuân thủ GMP của cơ sở sản xuất tính tại thời điểm kiểm tra thường kỳ gần nhất của cơ sở. Việc ước tính nguy cơ này dựa trên số lượng các điểm tồn tại được xác định từ lần kiểm tra gần nhất.
Sau khi đánh giá các nguy cơ nội tại và nguy cơ liên quan tới mức độ tuân thủ GMP của cơ sở (thực hiện sau khi hoàn thiện việc kiểm tra GMP), việc phối hợp hai nguy cơ này thông qua ma trận tối giản sẽ đưa ra được xếp loại nguy cơ của cơ sở sản xuất và tần suất kiểm tra giám sát tuân thủ GMP. Bảng chấm điểm các nguy cơ và hướng dẫn cách xếp loại nguy cơ nội tại và nguy cơ liên quan tới việc tuân thủ GMP được hướng dẫn tại Phụ lục II.
6.6. Kế hoạch kiểm tra hàng năm
- Tháng 12 hàng năm, Trưởng PhòngQLCL thuốc phân công chuyên viên đầu mối xây dựng kế hoạch kiểm tra trong năm kế tiếp. Trưởng phòng rà soát và đưa nội dung này bổ sung vào kế hoạch hoạt động hàng năm của Phòng, theo biểu mẫu BM.CL.01/14 – Kế hoạch kiểm tra GMP hàng năm.
- Kế hoạch kiểm tra hàng năm bao gồm:
+ Kiểm tra thường kỳ: trên cơ sở tần suất kiểm tra tái cấp chứng chỉ GMP là 3 năm.
+ Kế hoạch kiểm tra giám sát tuân thủ GMP: được xây dựng dựa trên xếp loại nguy cơ và tần suất kiểm tra giám sát tuân thủ GMP của các cơ sở sản xuất, thông tin về chất lượng thuốc, phản ứng ADR của thuốc
6.7. Kiểm soát thay đổi
Các cơ sở sản xuất thuốc, trong thời hạn chứng nhận GMP còn hiệu lực, phải có văn bản báo cáo trong các trường hợp sau:
a) Sửa chữa, thay đổi cấu trúc, sơ đồ bố trí nhà xưởng, dây chuyền sản xuất;
b) Thay đổi các thiết bị sản xuất chính;
c) Thay đổi lớn các hệ thống tiện ích ảnh hưởng môi trường sản xuất hoặc bản thân hệ thống: thay đổi nguyên lý thiết kế, vận hành....
d) Thay đổi người chịu trách nhiệm chuyên môn;
Cơ sở sản xuất vắc xin, sinh phẩm y tế, trong thời hạn giấy chứng nhận GMP còn hiệu lực, phải có văn bản báo cáo, trước khi tiến hành thay đổi trong các trường hợp sau:
a) Các trường hợp a, b, c như cơ sở sản xuất thuốc;
b) Sản xuất, sản xuất thử vắc xin hoặc sản phẩm khác trên dây chuyền sản xuất vắc xin đã được cấp chứng nhận.
c) Thay đổi lớn về quy trình sản xuất, kiểm tra chất lượng các sản phẩm vắc xin, sinh phẩm y tế.
Kèm theo báo cáo là bản đánh giá về nguy cơ, ảnh hưởng của các thay đổi dự kiến thực hiện đến chất lượng, an toàn của sản phẩm và các biện pháp áp dụng để giảm thiểu các nguy cơ, cùng với đánh giá hiệu quả của các biện pháp đó.
7. LƯU TRỮ HỒ SƠ TÀI LIỆU
Toàn bộ hồ sơ của Quy trình được bảo quản và lưu giữ theo quy định chung của Cục Quản lý Dược bao gồm:
- Hồ sơ đăng ký kiểm tra GMP của Cơ sở;
- Biên bản thẩm định hồ sơ đăng ký;
- Quyết định thành lập đoàn kiểm tra;
- Chương trình kiểm tra;
- Biên bản kiểm tra GMP;
- Báo cáo CAPA của Cơ sở;
- Biên bản đánh giá báo cáo CAPA;
- Phiếu trình Lãnh đạo Cục về việc cấp chứng chỉ GMP;
- Quyết định cấp giấy chứng nhận;
- Giấy chứng nhận GMP;
- Quyết định ngừng sản xuất đối với cơ sở không đạt GMP;
- Bản đánh giá đánh giá nguy cơ của cơ sở sản xuất
Các văn bản trong hồ sơ được đánh mã nhận dạng và nhóm lại theo từng bộ hồ sơ trong kẹp file. Mỗi hồ sơ có một checklist các văn bản thành phần của hồ sơ. Danh mục hồ sơ được cập nhật trên file mềm để tiện tra cứu.










